
GDC Query Filters for RNA-Seq Data
Data Operations, GDC Utilities 06/22/2024 OmixHub Team
Learn how to use GDC Query Filters to efficiently retrieve RNA-Seq data from the Genomic Data Commons (GDC) API.
Continue Reading...
Data Operations, GDC Utilities 06/22/2024 OmixHub Team
Learn how to use GDC Query Filters to efficiently retrieve RNA-Seq data from the Genomic Data Commons (GDC) API.
Continue Reading...
Data Operations, Google Cloud Utilities 05/15/2024 OmixHub Team
A comprehensive guide on uploading RNA-Seq expression data from GDC to your BigQuery database for efficient storage and analysis.
Continue Reading...
Data Operations, Data Preprocessing 04/03/2024 OmixHub Team
Learn how to create data matrices for Differential Gene Expression (DE) or Machine Learning analysis from GDC RNA-Seq data.
Continue Reading...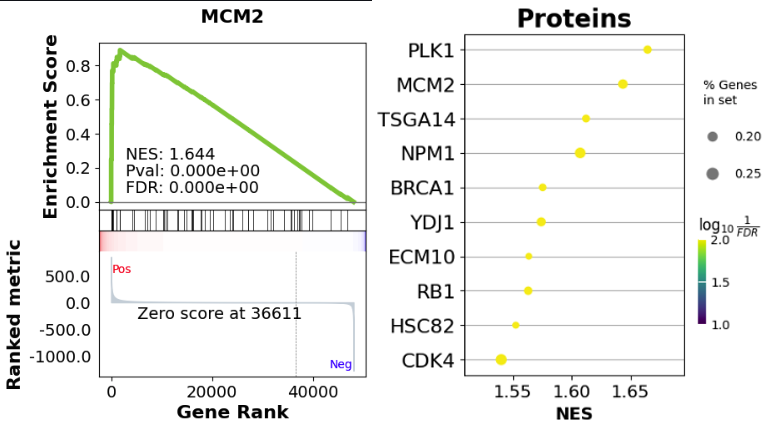
Data Operations, Feature Selection 03/01/2024 OmixHub Team
Explore techniques for selecting relevant features from high-dimensional RNA-Seq data to improve analysis and model performance.
Continue Reading...The Normal Tissue Sample Simulator is a powerful tool for balancing datasets with uneven distribution of normal and tumor samples. It uses an autoencoder to generate synthetic normal tissue samples, ensuring a more robust analysis.
This tool is particularly useful when:
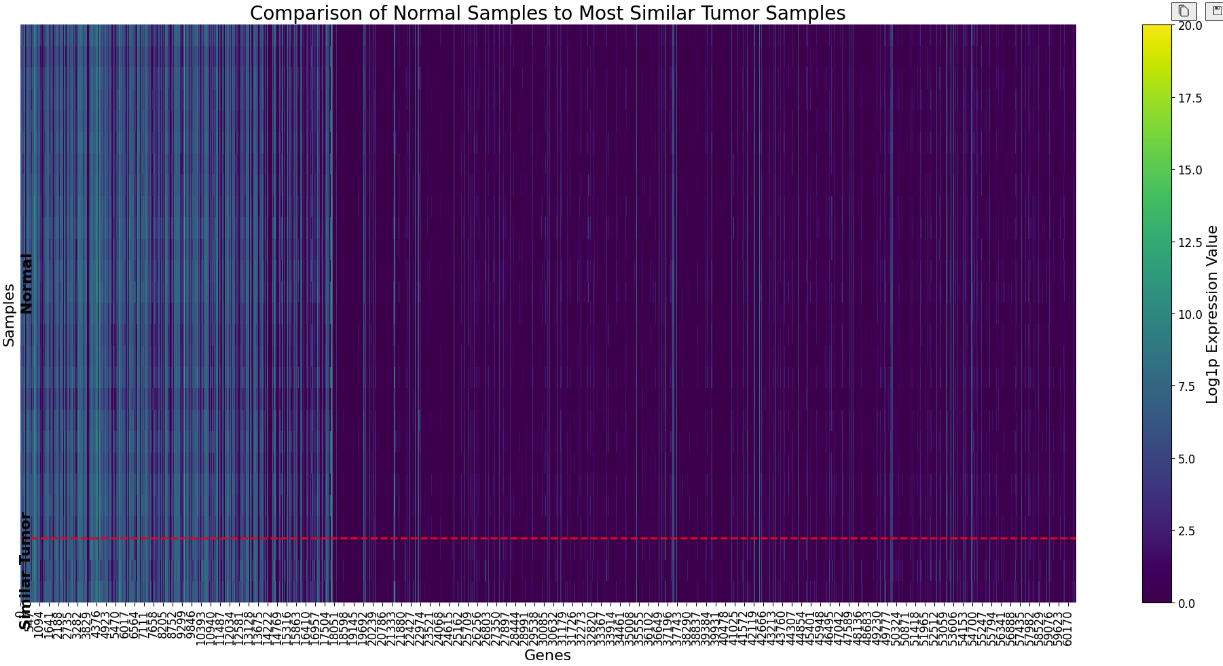
The figure above shows a heatmap plot comparing the distribution of original normal tissue samples with the simulated samples across 60K gene expression features. This visualization helps to verify that the simulated samples maintain the characteristics of the original normal tissue samples.
The Genomic Data Commons (GDC) provides a wealth of RNA-Seq data, but efficiently querying this data can be challenging. Our GDC Query Filters utility simplifies this process:
from Connectors.gdc_filters import GDCQueryFilters
gdc_filters = GDCQueryFilters()
rna_seq_filter = gdc_filters.rna_seq_filter()
# Use this filter with the 'files' endpoint
# Example: requests.post("https://api.gdc.cancer.gov/files", json={"filters": rna_seq_filter, "size": 10})
This utility allows you to create filters for various GDC data types and endpoints, making it easier to retrieve the specific data you need for your analysis.
Storing and analyzing large-scale RNA-Seq data can be challenging. Our Google Cloud utility helps you migrate GDC RNA-Seq expression data to BigQuery for efficient storage and analysis:
from Connectors.gcp_bigquery_utils import BigQueryUtils
from Engines.gdc_engine import GDCEngine
# Initialize BigQueryUtils and create table
bq_utils = BigQueryUtils(project_id='your_project_id')
table_id = 'your_project.dataset.table'
bq_utils.create_bigquery_table_with_schema(table_id, schema, partition_field="group_identifier", clustering_fields=["primary_site", "tissue_type"])
# Initialize GDCEngine and fetch data
gdc_eng_inst = GDCEngine(**params)
# Process and upload data for each primary site
for site in primary_sites:
json_object = gdc_eng_inst.make_count_data_for_bq(site, downstream_analysis='DE', format='json')
bq_utils.load_json_data(json_object, schema, table_id)
This approach allows for efficient uploading of data from multiple primary sites into a single, well-structured BigQuery table, optimized for query performance.
Preparing RNA-Seq data for downstream analysis is a crucial step. Our data preprocessing utility helps create cohorts for Differential Gene Expression (DE) or Machine Learning (ML) analysis:
import src.Engines.gdc_engine as gdc_engine
# Create Dataset for differential gene expression
rna_seq_DGE_data = gdc_eng_inst.run_rna_seq_data_matrix_creation(primary_site='Kidney', downstream_analysis='DE')
# Create Dataset for machine learning analysis
rna_seq_ML_data = gdc_eng_inst.run_rna_seq_data_matrix_creation(primary_site='Kidney', downstream_analysis='ML')
This utility allows you to easily create data matrices tailored for your specific analysis needs, whether it's for DE or ML studies.
Feature selection is crucial for improving model performance and interpretability in RNA-Seq data analysis. Our feature selection utility helps identify the most relevant genes:
import src.Engines.analysis_engine as analysis_engine
# Initialize the Analysis Engine
analysis_eng = analysis_engine.AnalysisEngine(data_from_bq, analysis_type='DE')
# Run differential expression analysis
res_pydeseq = analysis_eng.run_pydeseq(metadata=metadata, counts=counts_for_de)
# Perform Gene Set Enrichment Analysis (GSEA)
gene_set = 'Human_Gene_Atlas'
result, plot = analysis_eng.run_gsea(res_pydeseq_with_gene_names, gene_set)
This utility combines differential expression analysis with Gene Set Enrichment Analysis to help you identify the most important features (genes) for your RNA-Seq data analysis.
Here's how you can use the AutoencoderSimulator in conjuction with optimal transport to generate synthetic normal tissue samples, balancing your dataset for more accurate analysis:
# Get original normal samples
normal_samples = data_from_bq[data_from_bq['tissue_type'] == 'Normal']
# Simulate normal samples
simulator = simulators.AutoencoderSimulator(data_from_bq)
preprocessed_data = simulator.preprocess_data()
simulator.train_autoencoder(preprocessed_data)
num_samples_to_simulate = len(tumor_samples) - len(normal_samples)
simulated_normal_samples = simulator.simulate_samples(num_samples_to_simulate)
This code snippet demonstrates the process of:
After running this code, you can combine the original and simulated normal samples with your tumor samples to create a balanced dataset for further analysis.
Ask the OmixHub LLM Agent about data operations or to perform tasks: